DNA Is a Puzzle Enthusiast

Abstract
This article presents a concise summary of research projects in which Roberto Grossi participated, yielding interesting results that were never previously published in research papers. At the time, these studies were deemed too limited and in need of further extensions and generalizations, which were never realized due to a lack of resources. The researches focused on methods for inferring possible three-dimensional DNA conformations based on nucleotide sequence characteristics. Specifically, two key approaches were investigated: the identification of structured motifs for detecting Transcription Factor Binding Sites (TFBS) and the study of nested permutations using PQ-trees. This article describes the obtained results in selected case studies, their potential implications, and the current state of the art in these research areas.
Keywords and phrases:
DNA, 3D structure, PQ trees, structural motifsCategory:
ResearchCopyright and License:
2012 ACM Subject Classification:
Applied computing Bioinformatics ; Theory of computation Design and analysis of algorithmsEditors:
Alessio Conte, Andrea Marino, Giovanna Rosone, and Jeffrey Scott VitterSeries and Publisher:
1 Introduction
From a biochemical perspective, DNA is a polymeric macromolecule composed of four fundamental nucleotides: Adenine, Cytosine, Guanine, and Thymine. The genetic information encoded in DNA is determined by the specific sequence of these nucleotides. A well-known characteristic of DNA is its bilinear structure, consisting of two antiparallel strands. The chemical bonds linking the nucleotides are directional, giving each strand a specific orientation. Complementary base pairing follows strict rules: Adenine pairs with Thymine, while Cytosine pairs with Guanine.
The structural organization of DNA was first described in 1953 through the “double helix” model proposed by Watson and Crick, based on Rosalind Franklin’s X-ray diffraction data. This model, referred to as the “B-form double helix,” represents one of several possible DNA conformations. In fact, in living cells, DNA can assume more complex three-dimensional conformations, including single-, double-, triple-, and even quadruple-stranded regions [19]. Additionally, local complex structures, such as single-hairpin loops, cruciform DNA (double-hairpin structures), and other intricate conformations, have been observed [20].
Double-helix conformations result from the planar pairing of complementary bases between the two strands. More complex structures emerge from non-trivial base pairings within the same strand or between different strands. For example, if two sequences on the same strand are mutually inverted complemented, their pairing may generate cruciform (double-hairpin) structure (see Figure 1 for a schematic representation).
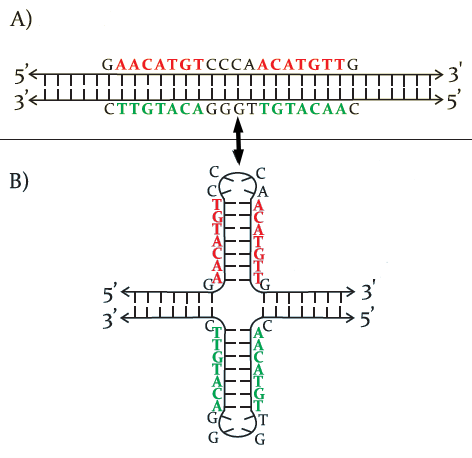
The study of DNA’s three-dimensional structures is crucial because, while the primary sequence encodes genetic information, its expression is mediated by proteins such as polymerases, transcription factors, and gene inhibitors and others. These proteins recognize specific DNA conformations rather than nucleotide sequences themselves. Molecular interactions, in fact, rely on “tactile” recognition, requiring precise surface contact to explicate biological functions. For example, a transcription factor may specifically recognize a hairpin structure with a defined size and folding pattern, and such a 3D structure is generated by a specific paring pattern of the nucleotides.
In bioinformatics applications, DNA is often represented as a string in the alphabet {A, T, C, G}, corresponding to the primary sequence of one strand, from which the complementary strand can be inferred relying on pairing rules. This representation is primarily used to characterize the informational content of DNA, but it is also indirectly linked to possible local geometries, given that these geometries arise from non-trivial base pairings within the same strand or between different strands.
Bioinformatics research has frequently focused on comparing DNA sequences, identifying similarities, character substitutions, insertions, deletions, and even more significant modifications at the chromosomal level. Various algorithms have been proposed (and continue to be developed) for comparing genomic sequences to detect differences between healthy individuals and those affected by specific diseases, reconstruct the phylogenetic history of evolutionarily related species, and support numerous other applications that have become common with the rise of omics sciences.
Attempts to investigate possible local structures based on the arrangement of letters within DNA sequences have been relatively more limited. Identifying repetitions, palindromes, inversions, and complementations with translocations, for example, provides a means to infer structures with probable functional significance. Roberto Grossi and Nadia Pisanti have made a significant contribution to this type of investigation, along with their collaborators. In particular, two biologically important directions of research have emerged: the identification of structured motifs [15, 10, 9, 4, 16, 17] and the representation of DNA’s primary sequence using PQ-trees, an efficient data structure that can identify sequences derived from each another through character permutations [3, 5, 7, 11].
2 Structured motifs in the DNA
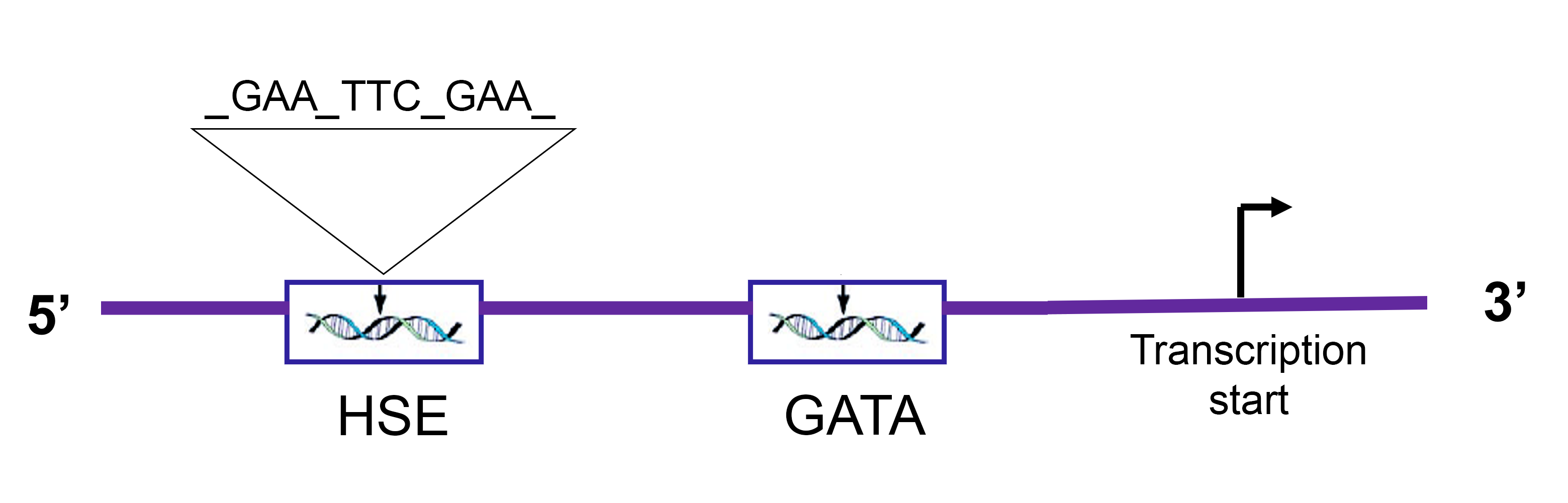
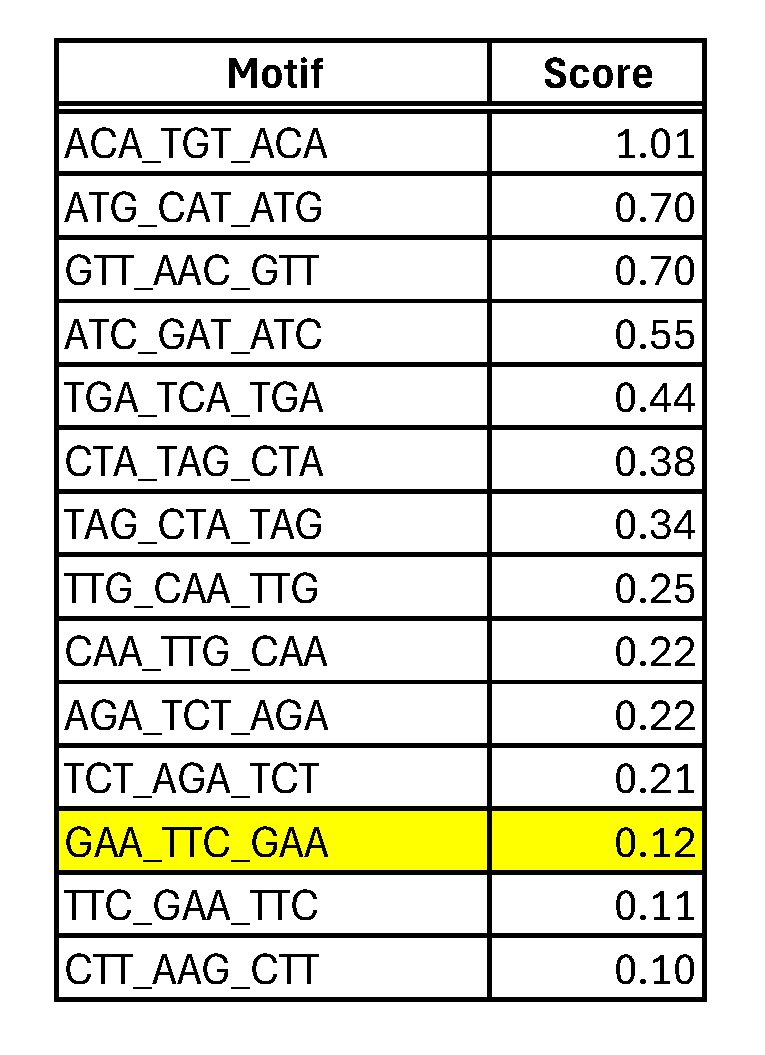
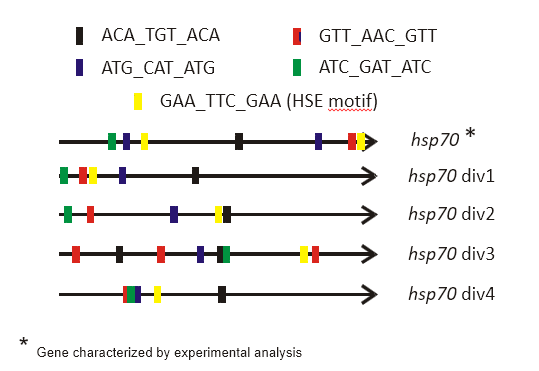
A case study of structural investigation by motifs search concerned the heat-shock genes activation in Tetrahymena termophila. Heat shock genes are a class of genes that are usually activated when a thermal stress might damage the structure of cellular proteins. This class of genes includes functionally different molecular tools (mainly chaperonins: i.e. proteins that help other proteins to assume the correct folding), usually named after the molecular weight of their protein product. For example, the hsp70 genes refer to the heat shock gene subclass, the protein products of which weight about 70 kD. T. thermophila genome presents several (about 30) hsp70 genes, called isoforms, which are very similar each other, but not perfectly identical, and they may have a different regulation mechanism. The firstly studied copy, named hsp70_1 appears to be regulated through a complex mechanism involving two seemingly unrelated sequences [1]. The first regulatory element is a simple GATA tetra-nucleotide, while the second, named HSE (heat-shock enhancer), exhibiting a relatively more intricate structure. The HSE is composed of three blocks: the first block contains the GAA three-nucleotide, separated from the second block by an interval of two arbitrary nucleotides. The second block features the TTC three-nucleotide, which is the inverted complement of the first block. Following an interval of approximately eight arbitrary nucleotides, the third block contains again the same three-nucleotide of the first block (see Figure 2 for a schematic representation). This arrangement allows the second block to pair with both the first and third blocks, leading to the formation of two distinct double-hairpin structures: one in which the first and second blocks pair within the same strand, and another in which the pairing is established between the second and third block [1]. The HSE sequence can oscillate between three distinct conformations: (i) a linear conformation with no intra-strand base pairing, and (ii) two alternative conformations featuring double-hairpin structures. The proposed mechanism for gene expression regulation involves structural stabilization: the transcription factor is likely to bind to one of the double-hairpin conformations, thereby stabilizing the proximal DNA structure and facilitating RNA polymerase access to the operator site. The functional role of the HSE sequence has been experimentally validated [1]: site-directed mutagenesis studies have demonstrated a significant reduction in HSP70 gene expression upon disruption of the HSE motif. A key biological question arising from this discovery was whether other hsp70 gene copies exhibited analogous regulatory sequences. To address this, we developed a variant of the SMILE algorithm, named BioMotif [2], specifically designed for the identification and statistical assessment of structured sequences based on grammatical properties of the DNA sequence itself (an evolution of SMILE algorithm has been published thereafter with the name RISOTTO [15]). We systematically searched for sequences located within 500 nucleotides upstream of gene start sites, structured into three distinct boxes, each three nucleotides long, with the central box containing an inverted complement of the sequence found in both the first and third boxes. In essence, the objective was to identify sequences capable of forming double-hairpin structures within close genomic proximity, a key structural characteristic identified in the experimentally studied sequence. The search yielded statistically significant results, with identified sequences located near the transcription start sites of each hsp70 isoform. The Figure 3 illustrates the localization of all detected sequences. Notably, the experimentally validated sequence was also identified, despite not being explicitly provided to the search algorithm. This finding strengthens the hypothesis that these sequences may also play functional roles, potentially acting as regulatory elements for the expression of hsp70 isoforms.
At this stage, the investigation was handed over to experimental biologists for site-directed mutagenesis studies to assess the involvement of additional candidate regulatory sequences. However, the termination of the research project and the disbanding of the experimental team indefinitely postponed this experimental validation, which is the primary reason why these findings were never published.
Nonetheless, we emphasize that this type of investigation, though relatively underutilized in biological research, can provide far more informative and effective insights than usual consensus sequence searches, which rely on conservation of bases rather than structural and functional properties. The largest (and regularly updated: the last version is v.10, 2024) database of TFBS (Transcription Factor Binding Sites), Jaspar ([8], [18]), still encode the TFBS regions as consensus matrices. Jaspar does not include T. thermophila in its species’ collection, therefore it is not possible to use it for validating BioMotif output. We anyhow performed a search of them, to investigate their presence in other organisms. We firstly transformed the most significant BioMotif results into consensus matrices following two simple criteria: a) conserved nucleotides get 100% of the consensus column, the other bases get 0; b) in “don’t care” positions, each nucleotide gets 25%. We assign a length of 3 to each “don’t care” spacer between conserved sequences. These matrices have been assigned to Jaspar for searching.
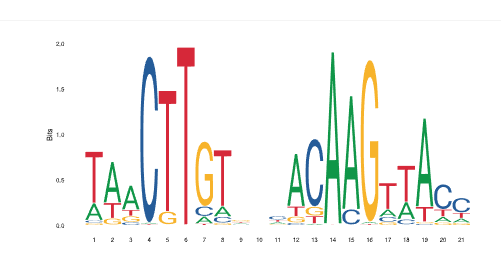
We have found no exact results in any taxonomic category, even when we searched for the experimentally discovered HSE motif, but this is not surprising, given that the Jaspar database does not contain T. thermopila, nor any evolutionary affine organism. But the search found several consensus sequences containing sub-strings of BioMotif results. For example, the search for the motif “CTT_AAG_CTT” (score = 0.10), returns a result in Arabidopsis thaliana, the consensus sequence of which is displayed in Figure 4. This is a TFBS of a NAC-gene family, involved in stress-responses [13], then with analogous function of hsp70 genes. The first two boxes of the query sequence, ”CTT_AAG”, are clearly present in it, but looking more in the detail, we find that the middle position of the first box shows the ”T” nucleotide with about 80% of conservation, while the other letter is an ”C”, with about 20%. A symmetric situation takes place for the second position in the second box: 80% ”A” and 20% ”G”. Jaspar does not give more details in summarized results, but this situation is compatible with the existence of 80% ”CTT_AAG”, and 20% ”CGT_ACG”: it is noticeable that both sequences are configuring the same structural motif: the second box is the complemented inverted of the first, thus probably giving rise to the same 3D conformation. In other words, it is reasonable to think that the 3D structure has been more preserved than the sequence itself: describing TFBS as structured motifs instead of consensus sequences could make this situation more explicit, with a significant improvement of the biological knowledge.
3 Searching for permutations using PQ trees
A further generalization beyond structured motifs consists of considering all possible permutations contained within, and often nested in, a DNA sequence. We regard this as a generalization because we do not predefine either a consensus sequence or a structured motif but instead investigate potential internal organizations within the sequence.
| PQ tree height | # trees |
|---|---|
| 1 | 18018 |
| 2 | 13583 |
| 3 | 10897 |
| 4 | 3412 |
| 5 | 3513 |
| 6 | 514 |
| 7 | 281 |
| 8 | 8 |
| 9 | 5 |
| 10 | 3 |
| 11 | 5 |
| 12 | 1 |
| 13 | 1 |
| 14 | 0 |
| 15 | 1 |
| 16 | 0 |
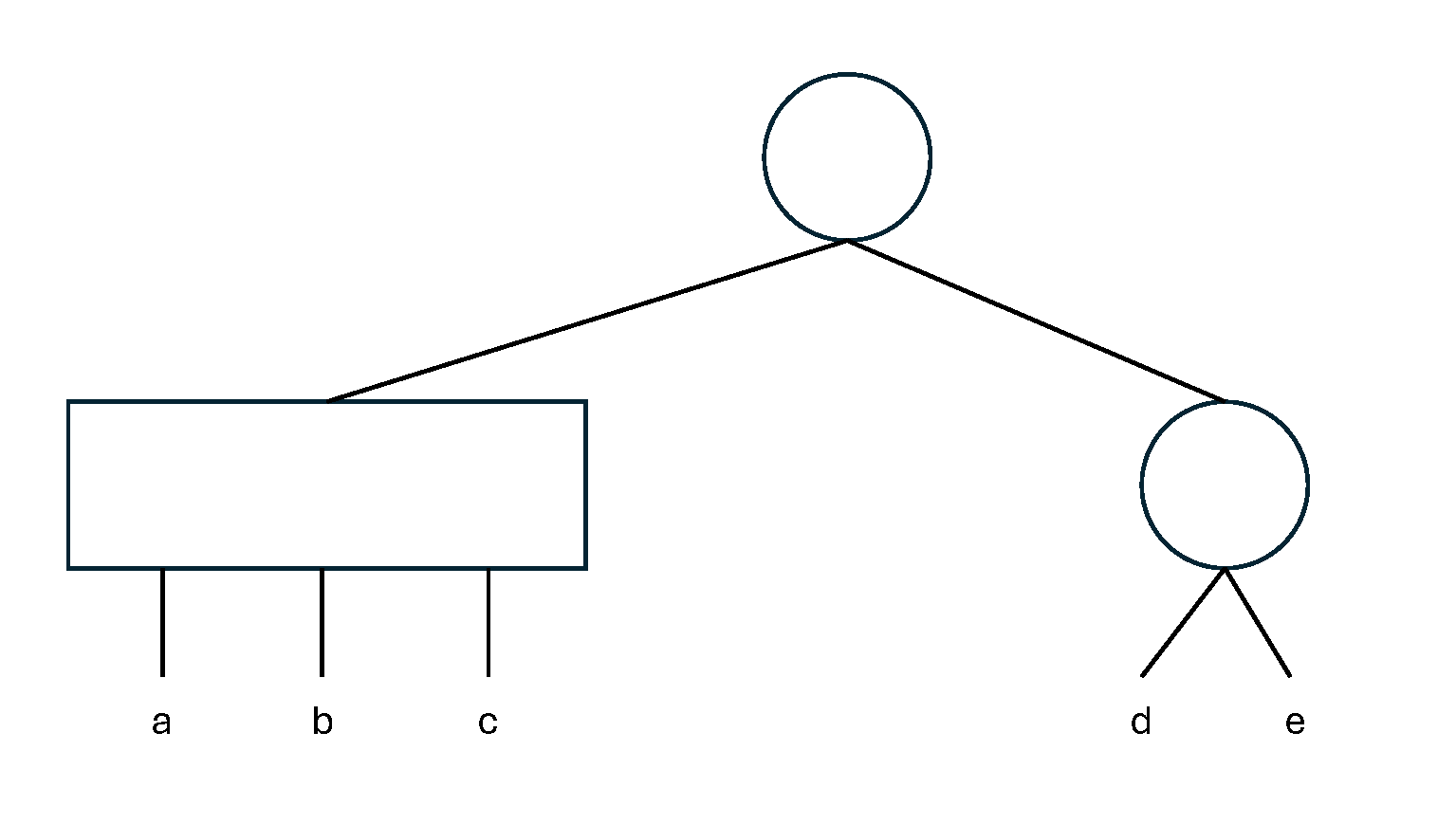
Permutations occurring at variable distances may give rise to a wide variety of 3D DNA conformations, which are probably not experimentally described but likely have functional significance. The primary data structure used in this research was the PQ tree, a notation introduced in the 1970s to represent maximal permutative structures within a sequence [6]. A PQ tree possesses two kind of nodes: type-P nodes (graphically represented by a circle), whose children can be permutated in any order, and type-Q nodes (graphically represented by a rectangle), whose chilren can occur only in the given order or in the reverse one, but not in other orders: Figure 5 presents an example of a PQ tree. A PQ tree is a natural way to represent a sequence containing substrings which are permutated each other, a very common situation for genomic DNA sequences. The application of this approach to biological sequences was the subject of two master’s theses, for which Roberto Grossi was among the advisors [7, 11]. From a computational perspective, these theses focused on the problem of efficiently generating the PQ tree representation of a given input sequence, minimizing both execution time and memory usage. The research led to the development of algorithms and procedures capable of extracting PQ trees in nearly linear time. Biological applications were implemented as simple test cases of the procedure. However, they yielded promising results that, unfortunately, were never further developed. These preliminary analyses were conducted on the gene encoding the principal glutamate receptor in Rattus norvegicus, called mGluR1, and accessible at ENA Nucleotide database at: https://www.ebi.ac.uk/ena/browser/view/M61099. The study did not aim to characterizing the generated PQ trees (they are too many, accounting for a huge number of permutations) but rather to examine the distribution of their height. The height of a PQ tree is, in practice, proportional by the degree of nesting of the permuted sequences, i.e. the number of times by which permutations of long regions contain, within them, permutations of shorter regions. One more nesting level increases the height of the relative PQ tree by 1; for example, the PQ tree in Figure 5 has height = 2. The Table 1 reports the number of PQ trees generated for each height. There are no trees with height , but there are several trees with height , thus showing a high-degree of nested permutations. This situation probably has functional significance, as a random shuffling of the sequence leads to a very different distribution, with a maximum height = 6 and not 15. In other words, the high level of permutation nesting is not random, but reasonably it is linked to some local arrangement of the DNA conformation. Most of the studies on PQ-trees and their potential applications in computational molecular biology were published between 2005 and 2012. In more recent years, only a few works have been published, with those exploring applications in biology being particularly scarce and almost exclusively focused on comparative genomics. Even recently, a PQ-tree structure has been proposed to address the problem of identifying and comparing gene clusters in bacterial genomes of strains/species closely related to already annotated genomes [14]. However, searches in SCOPUS bibliographic database have not retrieved any publication that investigates the height of PQ-trees to test its potential association to functional properties of genomic sequences.
4 Conclusions
We can conclude that there is an intriguing and still poorly investigated link between the various arrangements in the letters composing the DNA, when it is represented as a string, and the local conformation assumed by the DNA as a biological macromolecule. The presented approaches, structural motifs search and PQ-trees representation, are still valid and promising even though they have been proposed around 15-20 years ago. It is clear that DNA loves to play word games: it is surely a puzzle enthusiast and any approach that is able to establish a link between a peculiar arrangement of its letters and a biological function can produce very useful biological insights.
References
- [1] Sabrina Barchetta, Antonietta La Terza, Patrizia Ballarini, Sandra Pucciarelli, and Cristina Miceli. Combination of two regulatory elements in the tetrahymena thermophila HSP70-1 gene controls heat shock activation. Eukaryotic cell, 7(2):379–386, 2008.
- [2] Alessandro Bartolomei. BioMotif: un metodo per la ricerca di motivi altamente strutturati in sequenze genomiche. Master’s thesis, University of Pisa, IT, June 2007. Available at https://etd.adm.unipi.it/theses/available/etd-09252007-092605/unrestricted/Tesi.pdf.
- [3] Giovanni Battaglia. Discovery of unconventional patterns for sequence analysis: theory and algorithms. Phd thesis, University of Pisa, Italy, June 2011. Available at https://tesidottorato.depositolegale.it/handle/20.500.14242/128506. URL: https://etd.adm.unipi.it/theses/available/etd-12052011-215104/.
- [4] Giovanni Battaglia, Davide Cangelosi, Roberto Grossi, and Nadia Pisanti. Masking patterns in sequences: A new class of motif discovery with don’t cares. Theoretical Computer Science, 410(43):4327–4340, 2009. doi:10.1016/J.TCS.2009.07.014.
- [5] Giovanni Battaglia, Roberto Grossi, and Noemi Scutella. Consecutive ones property and PQ-trees for multisets: Hardness of counting their orderings. Information and Computation, 219:58–70, 2012. doi:10.1016/J.IC.2012.08.005.
- [6] Kellogg S Booth and George S Lueker. Testing for the consecutive ones property, interval graphs, and graph planarity using pq-tree algorithms. Journal of computer and system sciences, 13(3):335–379, 1976. doi:10.1016/S0022-0000(76)80045-1.
- [7] Giuseppe Camposeo. Scoperta di pattern ripetuti mediante l’uso di alberi pq. Master’s thesis, University of Pisa, IT, June 2007. Available at https://etd.adm.unipi.it/theses/available/etd-03132008-103339/unrestricted/Tesi.pdf.
- [8] Jaime A Castro-Mondragon, Rafael Riudavets-Puig, Ieva Rauluseviciute, Roza Berhanu Lemma, Laura Turchi, Romain Blanc-Mathieu, Jeremy Lucas, Paul Boddie, Aziz Khan, Nicolás Manosalva Pérez, et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic acids research, 50(D1):D165–D173, 2022.
- [9] Roberto Grossi, Giulia Menconi, Nadia Pisanti, Roberto Trani, and Søren Vind. Motif trie: An efficient text index for pattern discovery with don’t cares. Theoretical Computer Science, 710:74–87, 2018. doi:10.1016/J.TCS.2017.04.012.
- [10] Roberto Grossi, Andrea Pietracaprina, Nadia Pisanti, Geppino Pucci, Eli Upfal, and Fabio Vandin. MADMX: A strategy for maximal dense motif extraction. Journal of Computational Biology, 18(4):535–545, 2011. doi:10.1089/CMB.2010.0177.
- [11] Rosario Lombardo. Algoritmi efficienti per la scoperta di pattern ripetuti a intervalli. Master’s thesis, University of Pisa, IT, June 2008. Available at https://etd.adm.unipi.it/theses/available/etd-06182008-085952/unrestricted/Lombardo_2008__Laurea_Magistrale_in_Informatica.pdf.
- [12] SG Lushnikov, AV Dmitriev, Alexander Ivanovich Fedoseev, Gennady Aleksandrovich Zakharov, AV Zhuravlev, Anna Vladimirovna Medvedeva, BF Schegolev, and EV Savvateeva-Popova. Low-frequency dynamics of DNA in Brillouin light scattering spectra. JETP letters, 98:735–741, 2014.
- [13] Hisako Ooka, Kouji Satoh, Koji Doi, Toshifumi Nagata, Yasuhiro Otomo, Kazuo Murakami, Kenichi Matsubara, Naoki Osato, Jun Kawai, Piero Carninci, et al. Comprehensive analysis of NAC family genes in oryza sativa and arabidopsis thaliana. DNA research, 10(6):239–247, 2003.
- [14] Eden Ozeri, Meirav Zehavi, and Michal Ziv-Ukelson. New algorithms for structure informed genome rearrangement. Algorithms for Molecular Biology, 18(1):17, 2023. doi:10.1186/S13015-023-00239-X.
- [15] Nadia Pisanti, Alexandra M Carvalho, Laurent Marsan, and Marie-France Sagot. RISOTTO: fast extraction of motifs with mismatches. In LATIN 2006: Theoretical Informatics: 7th Latin American Symposium, Valdivia, Chile, March 20-24, 2006. Proceedings 7, pages 757–768. Springer, 2006. doi:10.1007/11682462_69.
- [16] Nadia Pisanti, Maxime Crochemore, Roberto Grossi, and M F Sagot. A basis of tiling motifs for generating repeated patterns and its complexity for higher quorum. In Mathematical Foundations of Computer Science 2003: 28th International Symposium, MFCS 2003, Bratislava, Slovakia, August 25-29, 2003. Proceedings 28, pages 622–631. Springer, 2003. doi:10.1007/978-3-540-45138-9_56.
- [17] Nadia Pisanti, Maxime Crochemore, Roberto Grossi, and Marie-France Sagot. Bases of motifs for generating repeated patterns with wild cards. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2(1):40–50, 2005. doi:10.1109/TCBB.2005.5.
- [18] Ieva Rauluseviciute, Rafael Riudavets-Puig, Romain Blanc-Mathieu, Jaime A Castro-Mondragon, Katalin Ferenc, Vipin Kumar, Roza Berhanu Lemma, Jérémy Lucas, Jeanne Chèneby, Damir Baranasic, et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic acids research, 52(D1):D174–D182, 2024.
- [19] Alexander Rich. DNA comes in many forms. Gene, 135(1-2):99–109, 1993.
- [20] Andrew Travers and Georgi Muskhelishvili. DNA structure and function. The FEBS journal, 282(12):2279–2295, 2015.